
Key Specs
-
All primers are collected from literature published in premier Microbiology journals within the past decade.
-
All primers are free to pick and ready for use in our laboratory at no cost to our customers. We also accept customerized primers.
-
The list covers both PE250/PE300 NGS pipeline and long-read PacBio pipeline.
Microbial Ribosomal RNA
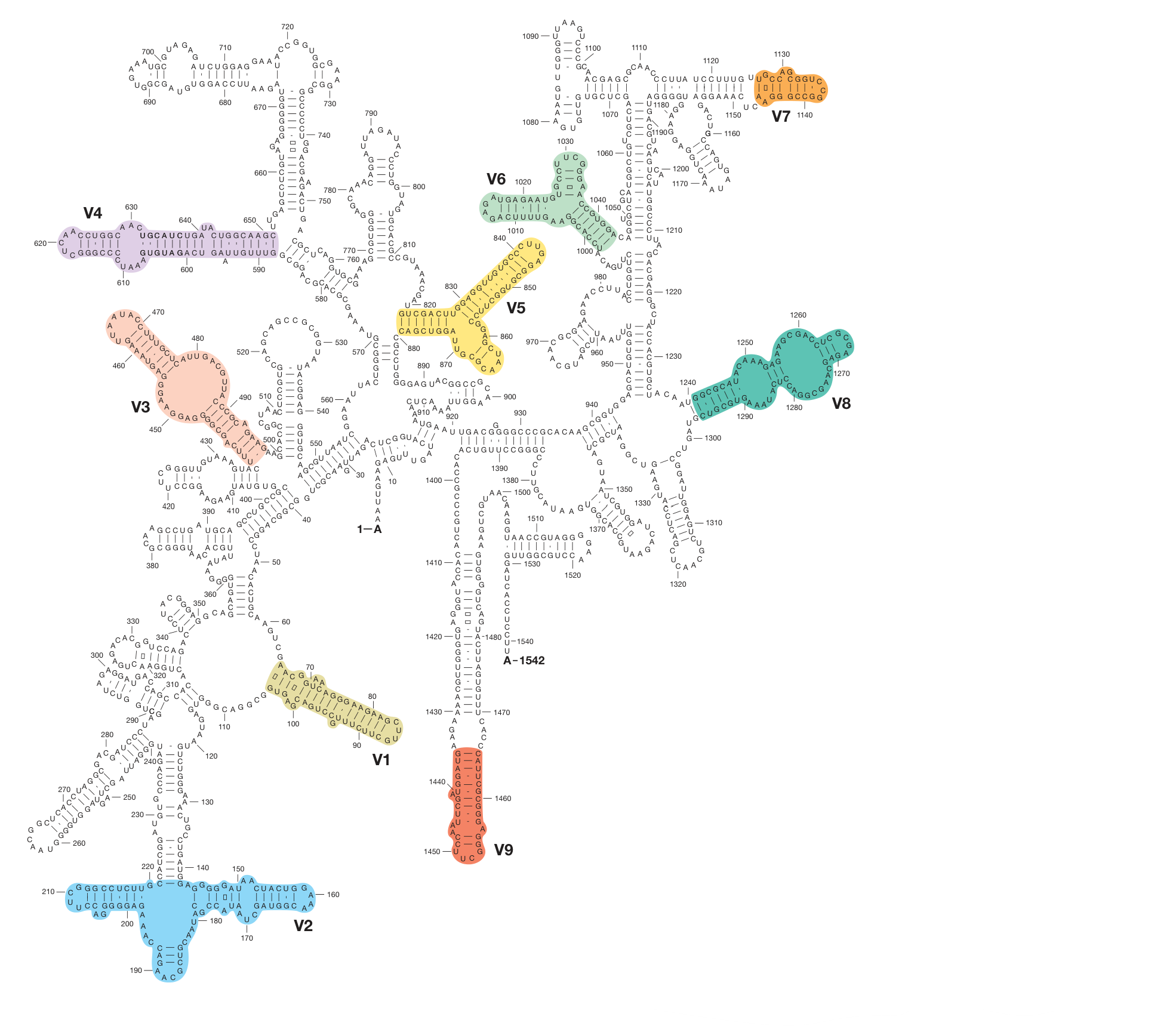
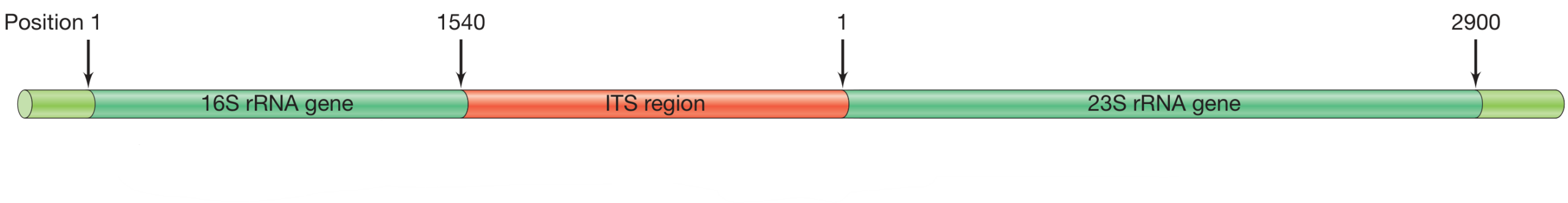
Primers (Short-read, NGS)
| Target | Primer | Primer Sequence | Amplicon length (bp) | PCR Region | Sequencing Strategy | Typical Target Groups |
| Bacterial 16S rRNA | 27F | 5′- AGAGTTTGATCCTGGCTCAG -3′ | ~311 | V1-V2 | PE250/PE300 | Human or animal microbiomes. |
| 338R | 5′- TGCTGCCTCCCGTAGGAGT -3′ | |||||
| Bacterial 16S rRNA | 27F | 5′- AGAGTTTGATCCTGGCTCAG -3′ | ~526 | V1-V3 | PE300 | Human or animal microbiomes. |
| 533R | 5′- TTACCGCGGCTGCTGGCAC -3′ | |||||
| Bacterial 16S rRNA | 515F | 5′-GTGCCAGCMGCCGCGG -3′ | ~392 | V4-V5 | PE250/PE300 | Environmental microbiomes. |
| 907R | 5′- CCGTCAATTCMTTTRAGTTT-3′ | |||||
| Bacterial 16S rRNA | 338F | 5′- ACTCCTACGGGAGGCAGCAG -3′ | ~468 | V3-V4 | PE250/PE300 | All types of bacterial communities. |
| 806R | 5′- GGACTACHVGGGTWTCTAAT -3′ | |||||
| Bacterial 16S rRNA | 341F | 5′- CCTAYGGGRBGCASCAG -3′ | ~466 | V3-V4 | PE250/PE300 | All types of bacterial communities. |
| 806R | 5′- GGACTACNNGGGTATCTAAT -3′ | |||||
| Bacterial 16S rRNA | 341F | 5′- CCTACGGGNGGCWGCAG-3′ | ~465 | V3-V4 | PE250/PE300 | Aquatic bacterial communities. |
| 785R | 5′-GACTACHVGGGTATCTAATCC-3′ | |||||
| Bacterial 16S rRNA | Ba9F | 5′-GAGTTTGATCMTGGCTCAG-3′ | ~506 | V1-V3 | PE300 | Rumen bacterial community in ruminants. |
| Ba515Rmod1R | 5′-CCGCGGCKGCTGGCAC-3′ | |||||
| Bacterial 16S rRNA | 343F | 5′- TACGGRAGGCAGCAG-3′ | ~456 | V3-V4 | PE250/PE300 | Human or animal microbiomes. |
| 798R | 5′- AGGGTATCTAATCCT-3′ | |||||
| Bacterial & Archaeal Universal 16S rRNA | 515FmodF | 5′-GTGYCAGCMGCCGCGGTAA-3′ | ~291 | V4 | PE250/PE300 | Recommended by the Earth Microbiome Project. |
| 806RmodR | 5′-GGACTACNVGGGTWTCTAAT-3′ | |||||
| Bacterial & Archaeal Universal 16S rRNA | 515F | 5′-GTGCCAGCMGCCGCGG-3′ | ~291 | V4 | PE250/PE300 | for both bacteria and archaea. |
| 806R | 5′-GGACTACHVGGGTWTCTAAT-3′ | |||||
| Bacterial & Archaeal Universal 16S rRNA | ArBa515F | 5′-GTGCCAGCMGCCGCGGTAA-3′ | ~250 | V4 | PE250/PE300 | for both bacteria and archaea with high coverage. |
| Arch806R | 5′-GGACTACVSGGGTATCTAAT-3′ | |||||
| Archaeal 16S rRNA | Arch344F | 5′-ACGGGGYGCAGCAGGCGCGA-3′ | ~571 | V3-V5 | PE300 | Archaeal communities. |
| Arch915R | 5′-GTGCTCCCCCGCCAATTCCT-3′ | |||||
| Archaeal 16S rRNA | 524F10extF | 5′-TGYCAGCCGCCGCGGTAA-3′ | ~434 | V4-V5 | PE250/PE300 | Commonly used for archaeal communities with high archaeal coverage. |
| Arch958RmodR | 5′-YCCGGCGTTGAVTCCAATT-3′ | |||||
| Archaeal 16S rRNA | Arch519F | 5′-CAGCCGCCGCGGTAA-3′ | ~400 | V4-V5 | PE250/PE300 | Commonly used for archaeal communities with high bacterial coverage. |
| Arch915R | 5′-GTGCTCCCCCGCCAATTCCT-3′ | |||||
| Archaeal 16S rRNA | Arch349F | 5′-GYGCASCAGKCGMGAAW-3′ | ~470 | V3V4 | PE250/PE300 | Commonly used for archaeal communities with high fungal coverage. |
| Arch806R | 5′-GGACTACVSGGGTATCTAAT-3′ | |||||
| Methanogenic Archaeal 16S rRNA | 1106F | 5′-TTWAGTCAGGCAACGAGC-3′ | ~273 | V9 | PE250/PE300 | Methanogens. |
| 1378R | 5′-TGTGCAAGGAGCAGGGAC-3′ | |||||
| Plant Endophytic 16S rRNA | 799F | 5′-AACMGGATTAGATACCCKG-3′ | ~450 | V5-V6 | PE250/PE300 | Plant endophytes. |
| V6R | 5′-GGGTTGCGCTCGTTGCG-3′ | |||||
| Plant Endophytic 16S rRNA | 799F | 5′-AACMGGATTAGATACCCKG-3′ | ~315 | V5-V6 | PE250/PE300 | Plant endophytes. |
| 1115R | 5′-AGGGTTGCGCTCGTTG-3′ | |||||
| Fungal ITS | ITS1F | 5′-CTTGGTCATTTAGAGGAAGTAA-3′ | ~300 | ITS1 | PE250/PE300 | Recommended by the Earth Microbiome Project. |
| ITS2R | 5′-GCTGCGTTCTTCATCGATGC-3′ | |||||
| Fungal ITS | 1737F | 5′-GGAAGTAAAAGTCGTAACAAGG-3′ | ~300 | ITS1 | PE250/PE300 | All types of fungal communities. |
| 2043R | 5′-GCTGCGTTCTTCATCGATGC-3′ | |||||
| Fungal ITS | ITS3F | 5′-GCATCGATGAAGAACGCAGC-3′ | 200~500 | ITS2 | PE300 | All types of fungal communities plus eukaryotes. |
| ITS4R | 5′-TCCTCCGCTTATTGATATGC-3′ | |||||
| Fungal ITS | gITS7 | 5′-GTGARTCATCGARTCTTTG-3′ | 250-500 | ITS2 | PE300 | All types of fungal communities plus eukaryotes. |
| ITS4 | 5′-TCCTCCGCTTATTGATATGC-3′ | |||||
| Fungal ITS | 5.8S-FunF | 5′-AACTTTYRRCAAYGGATCWCT-3′ | 267-511 | ITS2 | PE300 | All types of fungal communities with less eukaryote coverage. |
| ITS4-FunR | 5′-AGCCTCCGCTTATTGATATGCTTAART-3′ | |||||
| Fungal 18S rRNA | SSU0817F | 5′-TTAGCATGGAATAATRRAATAGGA-3′ | ~422 | V5-V6 | PE250/PE300 | Mostly for fungal groups: Ascomycota, Basidiomycota, Chytridiomycota, and Zygomycota. |
| 1196R | 5′-TCTGGACCTGGTGAGTTTCC-3′ | |||||
| Eukaryotic 18S rRNA | Euk1391f | 5′-GTACACACCGCCCGTC-3′ | ~200 | V9 | PE250/PE300 | Recommended by the Earth Microbiome Project. |
| EukBr | 5′-TGATCCTTCTGCAGGTTCACCTAC-3′ | |||||
| Eukaryotic 18S rRNA | TAReuk454FWD1F | 5′-CCAGCASCYGCGGTAATTCC-3′ | ~380 | V4 | PE250/PE300 | Protists excl. excavates and microsporidia. |
| TAReukREV3R | 5′-ACTTTCGTTCTTGATYRA-3′ | |||||
| Eukaryotic 18S rRNA | V4_1f | 5′-CCAGCASCYGCGGTAATWCC-3′ | ~380 | V4 | PE250/PE300 | Protists with higher coverage. |
| TAReukREV3R | 5′-ACTTTCGTTCTTGATYRA-3′ | |||||
| Eukaryotic 18S rRNA | 3NDF | 5′-GGCAAGTCTGGTGCCAG-3′ | ~450 | V4 | PE250/PE300 | Aquatic eukaryotes. |
| V4-euk-R2R | 5′-ACGGTATCTRATCRTCTTCG-3′ | |||||
| Eukaryotic 18S rRNA | 3NDF | 5′-GGCAAGTCTGGTGCCAG-3′ | ~450 | V4 | PE250/PE300 | Aquatic eukaryotes. |
| V4-euk-R1R | 5′-GACTACGACGGTATCTRATCRTCTTCG-3′ | |||||
| Eukaryotic 18S rRNA | V4F | 5′-GCGGTAATTCCAGCTCCAATA-3′ | 300-450 | V4 | PE250/PE300 | Eukaryotic phytoplankton |
| V4R | 5′-GATCCCCHWACTTTCGTTCTTGA-3′ | |||||
| Eukaryotic 18S rRNA | 3NDF | 5′-GGCAAGTCTGGTGCCAG-3′ | ~570 | V4 | PE300 | Soil nematode |
| 1132rmodR | 5′-TCCGTCAATTYCTTTAAGT-3′ | |||||
| Eukaryotic 18S rRNA | 528F | 5′-GCGGTAATTCCAGCTCCAA-3′ | 244-302 | V4 | PE300 | Picoeukaryotes |
| 706R | 5′-AATCCRAGAATTTCACCTCT-3′ | |||||
| Eukaryotic 18S rRNA | 616F | 5′-TTAAARVGYTCGTAGTYG-3′ | ~530 | V4 | PE300 | Eukaryotes with high coverage. |
| 1132R | 5′-CCGTCAATTHCTTYAART-3′ | |||||
| Eukaryotic 18S rRNA | Euk528f | 5′-CCGCGGTAATTCCAGCTC-3′ | ~410 | V4 | PE250/PE300 | Eukaryotic green algae. |
| CHLOO2r | 5′-CTTCGAGCCCCCAACTTTC-3′ |
Primers (Full-length, PacBio)
| Target | Primer | Primer Sequence | Amplicon length (bp) | PCR Region | Sequencing Strategy | Typical Target Groups |
| 16S rRNA | 27F | 5′- AGRGTTYGATYMTGGCTCAG -3′ | ~1500 | V1-V9 | PacBio | All types of bacterial communities |
| 1492R | 5′- RGYTACCTTGTTACGACTT -3′ | |||||
| 16S rRNA | A27F | 5′- AGRGTTYGATYMTGGCTCAG -3′ | ~1500 | V1-V9 | PacBio | All types of bacterial communities |
| A1492R | 5′- TASGGHTACCTTGTTASGACTT -3′ | |||||
| Fungal ITS | ITS1F | 5′-CTTGGTCATTTAGAGGAAGTAA -3′ | 300-900 | ITS1-ITS2 | PacBio | All types of fungal communities |
| ITS4R | 5′- TCCTCCGCTTATTGATATGC-3′ | |||||
| Fungal ITS | ITS1FngsF | 5′-GGTCATTTAGAGGAAGTAA -3′ | 300-900 | ITS1-ITS2 | PacBio | Environmental and clinical fungi |
| ITS4ngsR | 5′- TCCTSCGCTTATTGATATGC-3′ | |||||
| Fungal ITS | ITS9MunngsF | 5′-TACACACCGCCCGTCG -3′ | 300-900 | ITS1-ITS2 | PacBio | Soil fungi |
| ITS4ngsR | 5′- TCCTSCGCTTATTGATATGC-3′ | |||||
| Eukaryotic 18S | EukA | 5′-AACCTGGTTGATCCTGCCAGT-3′ | 1800 | V1-V9 | PacBio | Environmental eukaryotic microbiomes |
| EukB | 5′-GATCCTTCTGCAGGTTCACCTAC-3′ |
Full Method
High-throughput 16S ribosomal RNA gene sequencing
Microbial 16S rRNA genes was amplified with the selected primer sets (see table above for commonly used primers). Both the forward and reverse primers were tailed with sample-specific Illumina index sequences to allow for deep NGS sequencing. The PCR was performed in a reaction mixture of DNA template 5-50 ng, 0.3 μL forward primer (10μM), 0.3 μL reverse primer (10μM), 5 μL Taq DNA Polymerase Buffer, 2 μL dNTP (2 mM each), 0.2 μL Taq DNA Polymerase (Takara Bio Europe), and finally ddH2O added to a total volume of 20 μL. After initial denaturation at 95 °C for 5 min, followed by 20 cycles of denaturation at 95 °C for 30 s, annealing at 50 °C for 30 s, and extension at 72 °C for 40 s, and a final step at 72 °C for 7 min. The amplified products were purified with an Omega DNA purification kit (Omega Inc., Norcross, GA, USA) and were quantified using Qsep-400 (BiOptic, Inc., New Taipei City, Taiwan). The insert sizes of sequencing libraries were inspected using a LabChip (PerkinElmer, USA). The amplicon library was paired-end sequenced (2×250) on an Illumina Novaseq 6000 sequencer.
For PacBio full-length 16S rRNA gene sequencing, microbial DNA was additionally subjected to single-molecule real-time (SMRT) sequencing using the PacBio platform. Nearly full-length 16S rRNA genes (~1.5 kb) were amplified using universal primers targeting the V1–V9 regions (e.g. 27F and 1492R), with PacBio-specific barcode sequences attached to both primers to allow sample multiplexing.
PCR amplification was carried out using a high-fidelity DNA polymerase to minimize amplification errors. Each reaction contained 5–20 ng of template DNA, 0.5 μM of each primer, dNTPs, polymerase buffer, and high-fidelity DNA polymerase in a total volume of 25 μL. Thermal cycling conditions included an initial denaturation at 95 °C for 3 min, followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 90 s, with a final extension at 72 °C for 10 min.
Amplicons were purified using AMPure PB magnetic beads (Pacific Biosciences, USA) and quantified using a Qubit fluorometer (Thermo Fisher Scientific). Equimolar amounts of purified amplicons were pooled to construct SMRTbell libraries according to the manufacturer’s protocol. Libraries were size-selected to enrich for full-length inserts and sequenced on the PacBio Sequel II platform using circular consensus sequencing (CCS) mode to generate high-accuracy HiFi reads.
NGS data pre-treatment
The quality of raw reads was assessed using the program Fastqc. Raw reads were first filtered by Trimmomatic (version 0.33) with parameters: window size of 50 bp and average Q-score within the window as min. 20. Identification and removal of primer sequences were processed by Cutadapt (version 1.9.1) with a maximum mismatch set as 20% and minimum coverage of 80%. PE reads were assembled by USEARCH (version 10) with a minimum length of overlap of 10 bp, minimum similarity within the overlapping region as 90%, and maximum mismatch accepted as 5 bp. The chimera removal was done using UCHIME (version 8.1). If a fragment with over 80% similarity to the query sequence was found on both parents, this query sequence was defined as a chimera sequence. The high-quality reads generated were used in the following analysis.
ASV analysis
DADA2 method in QIIME2 (version 2020.06) was used to de-noise sequences, generating ASVs. The conservative threshold for OTU filtration is 0.005%. Each ASV was annotated using a combination of BLAST-based method and a Naive Bayes classifier-based method. The BALST-based annotation method was done with the Classify-consensus-blast functionality in QIIME2, which identifies the annotation with the highest consensus in N best hits (default: 3). Parameter setting: minimum similarity in sequence: 90%; minimum coverage: 90%; minimum consensus: 51%. The Naive Bayes classifier-based method is processed with the classify-sklearn plugin in QIIME. The classifier needs to be trained before use in order to “learn” which features can be used for classification. Parameter setting: the confidence of classifier set as 0.7. In general, feature sequences are firstly BLASTed against the reference database (Silva) using classify-consensus-blast. The sequences that could not be matched in the reference database were further classified by classify-sklearn.
Alpha diversity
Alpha diversity was calculated using QIIME2 (https://qiime2.org/), which reflects the species richness of individual samples and the species diversity. Chao1 and Ace index measure species richness, i.e. the number of species. Shannon and Simpson’s indexes are used to measure species diversity, which is affected by species richness and community evenness in the sample community. In the case of the same species richness, the higher the evenness of each species in the community is, the higher the community diversity is. A larger Shannon index and a smaller Simpson index indicate that the species diversity of the sample is higher.
Beta diversity
Beta diversity analysis was processed using QIIME2 (https://qiime2.org/), with the aim to compare species diversity between different samples. There are four commonly used statistical algorithms to calculate the distance between samples in beta diversity analysis, that is binary Jaccard, Bray Curtis, weighted Unifrac, and unweighted Unifrac. These four algorithms can be classified into weighted (Bray-Curtis and Weighted Unifrac) and non-weighted (Jaccard and Unweighted Unifrac). The unweighted algorithm focuses on the existence of a species, while the weighted algorithm takes both existence and abundance into consideration.
The unweighted Pair-group Method with Arithmetic Mean (UPGMA) distance analysis method was used to construct a clustering tree based on the similarity between samples. Distance matrices were obtained by the four distance algorithms mentioned above. Hierarchical clustering of samples was performed by UPGMA in the R language tool to determine the similarity in species composition among samples.




