
Key Takeaways
16S rRNA genes have intragenomic heterogeneity which can lead to taxonomic misclassification and overestimation of microbial diversity.
The V4 to V5 introduces the lowest overestimation rate (4.4%) but exhibits slightly lower species resolution.
The threshold for full-length 16S rRNA genes is 98.5~99% to minimize the overestimation rate at the species level.
Title and Copyright Information

Attention: all answers provided are solely based on this research paper and thus please be mindful of any potential biases.
Pan et al., 2023. Microbial Diversity Biased Estimation Caused by Intragenomic Heterogeneity and Interspecific Conservation of 16S rRNA Genes. Applied and Environmental Microbiology. 89:5. https://doi.org/10.1128/aem.02108-22.
What is 16S rRNA gene?
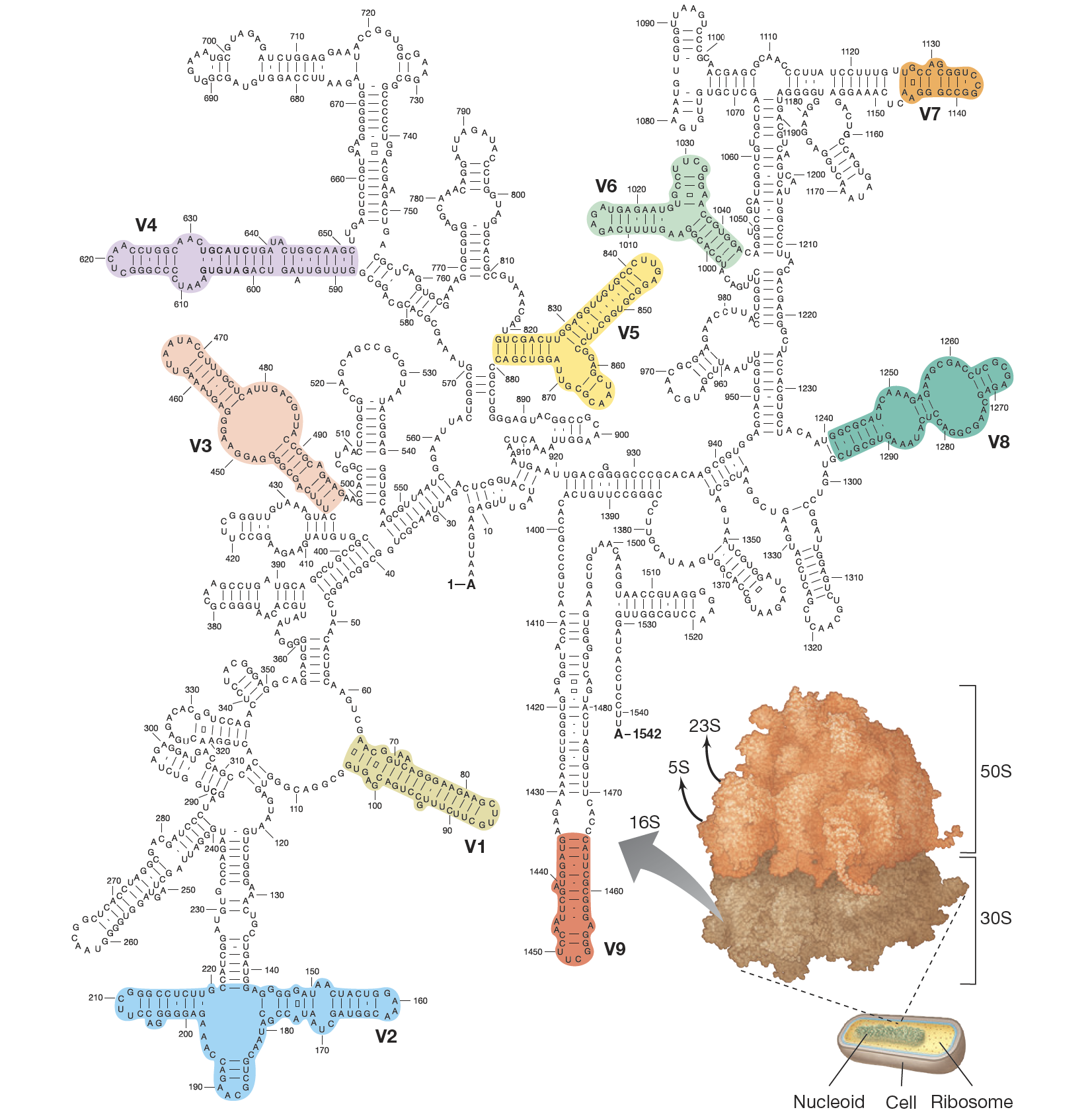
Figure Caption:
This is the primary and secondary structure of 16S rRNA from Escherichia coli (Bacteria). The 16S rRNA from Archaea is similar in secondary structure (folding) to that of Bacteria but has numerous differences in primary structure (sequence). The molecule is composed of conserved and variable regions (V1–V9). The approximate positions of the variable regions are indicated in color. Inset: the 70S ribosome of Bacteria is composed of 30S and 50S subunits; 16S rRNA is part of the 30S subunit whereas 5S and 23S rRNAs are parts of the 50S subunit.
Source:
Madigan, Michael T., Kelly S. Bender, Daniel H. Buckley, W. Matthew Sattley, and David A. Stahl. Brock Biology of Microorganisms. 16th ed. Pearson, 2021.
Why 16S rRNA gene?
The 16S rRNA gene has been widely adopted as a universal biomarker for microbial diversity studies because of some of its unique features.
- Ubiquity: The 16S rRNA gene is found in all bacteria and archaea, and even in eukaryotes within mitochondria and chloroplasts. This gene is highly conserved across different organisms, making it a suitable target for studying microbial diversity.
- Variability: Different regions of the 16S rRNA gene exhibit varying levels of variability. Some regions are highly conserved and similar in all organisms, while others are highly variable and differ greatly between distantly related organisms. This variability allows for distinguishing between different microbial taxa and provides valuable information for taxonomic classification.
- Phylogenetic Consistency: Phylogenies derived from the 16S rRNA gene generally agree with phylogenies derived from other conserved genes. This suggests that the evolutionary history of the organism as a whole can be represented by the 16S rRNA gene. Thus, it provides insights into the evolutionary relationships between different microbial taxa.
- Cultivation-Independent Methods: The 16S rRNA gene has been extensively studied using cultivation-independent methods, which do not rely on the cultivation of microorganisms in the laboratory. These methods have revolutionized the field of microbial ecology by enabling the study of unculturable or difficult-to-culture microorganisms.
- Technological Advances: The development of high-throughput sequencing (HTS) technologies has significantly increased the throughput and reduced the cost of sequencing. This has made it economically feasible for research groups to obtain rRNA gene data and explore microbial diversity. HTS has allowed for the generation of large numbers of sequences from multiple samples simultaneously, providing a more comprehensive understanding of microbial communities.
Cons:
- Limited Taxonomic Resolution: The 16S rRNA gene may not provide enough resolution to differentiate closely related species or strains. In some cases, additional genes or genomic regions may be required for more accurate identification.
- PCR Biases: The PCR amplification step introduces biases that can affect the representation of different microbial taxa in the final dataset. These biases can be influenced by primer selection, PCR conditions, and DNA extraction methods, potentially leading to an underrepresentation or overrepresentation of certain taxa.
- Incomplete Coverage: While the 16S rRNA gene is present in most bacteria and archaea, there are some exceptions. Some microbial groups may have variations in their 16S rRNA gene sequences that make them difficult to amplify or classify accurately.
- Lack of Functional Information: The 16S rRNA gene provides information about the phylogenetic relationships between microbial taxa but does not provide direct information about their functional capabilities or metabolic potential. Additional genomic or metagenomic analyses may be required to gain insights into the functional diversity of microbial communities.
Q&A
Q1: Please give a brief summary of this study and highlight the main discoveries.
The study conducted an analysis of 24,248 complete prokaryotic genomes to examine the bias in estimating microbial diversity caused by intragenomic heterogeneity and interspecific conservation of 16S rRNA genes. It was found that variable copy numbers, intragenomic heterogeneity, and low taxonomic resolution have caused biases in estimating microbial diversity. The results provide quantitative data on the degree of overestimation due to intragenomic heterogeneity and the extent of underestimation caused by the insufficient interspecific variation of 16S rRNA genes in prokaryotic genomes. The study also proposes the optimal identity thresholds for full-length and multiple partial 16S genes to minimize the risk of overmerging and over-splitting.
Q2: What are the average copy numbers of 16S rRNA gene in archaeal and bacterial genomes?
The average copy number of 16S rRNA genes in archaea was 1.7 ± 0.9, while in bacteria it was 5.3 ± 2.8. The mean copy number per phylum ranged between 1 and 6.9 ± 2.8 in bacteria and 1 and 2.0 ± 0.9 in archaea. The Firmicutes had the highest average copy number of 6.9 ± 2.8 in bacteria, followed by the Proteobacteria (5.5 ± 2.5) and Fusobacteria (5.2 ± 1.1). Low mean copy numbers were observed in the Acidobacteria (1.1 ± 0.3), Thermodesulfobacteria (1.1 ± 0.4), and Chloroflexi (1.4 ± 0.7) in bacteria, and the “Candidatus Korarchaeota”, “Candidatus Lokiarchaeota”, “Candidatus Micrarchaeota”, “Candidatus Nanohaloarchaeota”, and “Candidatus Thermoplasmatota” in archaea.
Q3: Compare the strategy of using full-length vs partial 16S rRNA gene for profiling microbial diversity.
The study of 16S rRNA gene has revealed that the intragenomic heterogeneity and the low taxonomic resolution of 16S rRNA genes introduce various degrees of bias in the estimation of microbial diversity. Analysis of the bias in estimating such prokaryotic diversity using 16S rRNA genes within prokaryotic genomes has highlighted the importance of the choice of sequencing regions and clustering thresholds based on the specific research objectives.
The V4 to V5 region was proposed as the optimal region for 16S rRNA gene-based microbial analyses due to its least intragenomic variation, while full-length 16S rRNA gene has higher thresholds for species delineation such as 98.5%, 98.65%, and 98.7 to 99.0%. Results of the study indicate that the optimal identity thresholds for full-length and multiple partial 16S genes are 98.7 to 99.0% and 97% respectively, to minimize the risk of overmerging and over-splitting. Thus, for profiling microbial diversity, it is recommended to use the V4-V5 region with 97% identity threshold, or full-length 16S rRNA gene with 98.7 to 99.0% identity threshold.
The choice of full-length or partial 16S rRNA sequencing should be based on the specific research objectives. Full-length 16S rRNA gene sequencing can lead to higher overestimation of microbial diversity compared to partial regions, due to intragenomic variation and heterogeneity. When using full-length 16S rRNA gene sequencing, an overestimation of 156.5% was measured at the ASV level. Generally, a low identity threshold (e.g., 97%) may cluster sequences from different species or even genera into the same OTU, while a too high threshold is likely to split a species or even a strain into multiple distinct clusters due to intragenomic variation. The optimal identity thresholds of species delimitation for full-length and partial 16S genes should be determined separately according to the specific research objectives, and the appropriate thresholds for different amplified fragments should be determined separately rather than depending on general experience.
Q4: What is the advantages and disadvantages of using full-length over partial 16S rRNA gene for profiling microbial diversity?
Full-length 16S rRNA gene has a higher interspecific conservation, thus providing better resolution for species delineation; however, it is prone to intragenomic heterogeneity and overestimation of microbial diversity, as revealed in a comprehensive examination of bias in estimating such prokaryotic diversity using 16S rRNA genes within prokaryotic genomes. Meanwhile, when using partial 16S genes, the risk of overestimation is much lower and the amplicon analysis based on the V3 to V4 region may only cause an overestimation of 51.7% for microbial diversity. However, the interspecific conservation of partial 16S rRNA gene is weaker than that of full-length gene, thus resulting in underestimation of the extant prokaryotic diversity.
Q5: What are OTU and ASV and which one is better for measuring microbial diversity?
OTU and ASV are both used to calculate microbial diversity. OTU (Operational Taxonomic Unit) is a cluster of sequences that are highly similar to each other, with the similarity threshold usually set at 97% or higher. ASV (Amplicon Sequence Variants) is a more recent method which uses a higher similarity threshold (99%-100%) to avoid overmerging caused by intragenomic variation and hence provides a more accurate estimation of microbial diversity. Compared to OTU, ASV can better differentiate closely related microbial species while maintaining the same taxonomic resolution. However, OTU is more commonly used due to its higher computational efficiency and lower data requirements.
Q6: What is the best strategy for profiling microbial diversity at the species level?
The best strategy for profiling microbial diversity at the species level is to use full-length 16S rRNA gene sequencing, which can provide a more accurate estimation of microbial diversity than the commonly used V4 to V5 region. Clustering of sequences should be done using identity thresholds between 98.5-99.0% to minimize the risk of overmerging and oversplitting. The NCBI or GTDB taxonomy should be used for the analysis.




